
In our introduction to cholesterol article, we discussed the basics of cholesterol and blood test interpretation. We also covered the major factors that influence blood cholesterol levels to build a strategy for managing high blood cholesterol. Our discussion has been highly simplified for practical purposes, but the relationship between diet, blood cholesterol levels, and the risk of disease is very complex.
This complexity causes a lot of confusion and misinformation to spread on the internet and in the media. Let’s address some of the most common concerns and criticisms. As a warning, this piece will be lengthy. It can be used as a reference on a case-by-case basis for each claim, since each section is written to be read in stand-alone fashion, or it can be read straight through for the motivated reader. These discussions will also involve more complex language and scientific discussion compared to our introductory guide.
Table of Contents
2 It’s not the cholesterol, it’s the inflammation. 3
4 Whether you have large or small LDL particles is more important than the number. 7
5 This study shows statins don’t work in the elderly. 8
6 This study found that lots of patients who have heart attacks had normal cholesterol levels. 9
7 If your Coronary Artery Calcium score is zero there’s nothing to worry about. 12
8 Statins cause lots of problems and you’ll lose all your gains. 14
9 This study found no relationship between high vs. low saturated fat intake and heart disease. 16
9.2 Food Sources & Replacement Effects. 19
11 How do you explain the increase in rates of heart disease deaths?. 24
*The above list is clickable and will take you to the listed section of the article.
1 Should we really be lowering blood cholesterol? Why would we make it if it causes these problems?
Cholesterol plays many important roles in the body. Many people assume that blood levels reflect whether we have enough cholesterol available to perform essential tasks like cell function, hormone synthesis (including testosterone), and many others. This is not true. Every cell can make essentially all the cholesterol it needs without relying on high amounts to be delivered from the bloodstream. A cholesterol test measures the amount of cholesterol being carried on lipoproteins in the blood. It does not measure how much cholesterol is inside our cells and tissues. This means your blood test is not telling you how much cholesterol your cells have at all, or how they are using cholesterol for normal body functions.
During early life when tissues are growing and cholesterol needs are at their highest, typical low-density lipoprotein-cholesterol (LDL-C) levels in human blood are only around 30 mg/dL. Brown 1986 Isolated hunter-gatherer societies maintain blood LDL-cholesterol levels in this range of 35-70 mg/dL throughout life without complications and show very low rates of heart disease across the lifespan. This suggests that these are likely normal levels for humans, contrasted against average levels in modern Americans closer to 130 mg/dL. O’Keefe 2004 Even in people who do not have any other conventional risk factors for heart disease like diabetes, high blood pressure, smoking, or obesity, LDL-cholesterol still has a linear, independent relationship with plaque development, starting from about 70 mg/dL on up. Friera 2017 We also know that lowering blood LDL-cholesterol to less than 70 mg/dL can halt the progression of plaque. Ahmadi 2019 In other words, even in the supposedly normal range of cholesterol, as levels increase further, so too does the risk of developing plaque. We will return to this question of what we should consider “normal” cholesterol levels in Section 8.
Blinded, placebo-controlled studies of medications in individuals at extremely high risk of heart disease have achieved extremely low blood LDL-cholesterol levels, down to 6.9 mg/dL. Giugliano 2017 Robinson 2017 This lowered heart disease risk without increasing the incidence of death, cancer, cognitive decline, muscle problems, hypogonadism (clinically significant low testosterone), sexual problems, or infertility. It is also notable that many of these medications work by increasing the expression of LDL receptors, which increases the rate that LDL particles are removed from the blood into the tissues.
Testosterone production is a common concern among athletes and lifters. It is true that testosterone production requires cholesterol. Recall that our cells make essentially all the cholesterol they need without relying on large amounts to be delivered from the blood. Additionally, blood cholesterol tests do not tell us anything about the amount of cholesterol being created and used inside of our cells. This is relevant where testosterone is synthesized in the testes. The testes rely heavily on their own synthesized cholesterol to create testosterone, not cholesterol delivered from the blood. Hu 2010 As a result, it is unsurprising that despite common concerns among athletes and lifters that lowering blood LDL-cholesterol levels might impair testosterone production, the evidence on medications used for this purpose do not show a higher risk of clinical hypogonadism (i.e., significant “low testosterone”), or any significant changes in the hormones that “sense” and regulate blood testosterone levels (follicle stimulating hormone and luteinizing hormone). Stamerra 2021 Travia 1995 Dobs 2009 Mondul 2010
We do not find direct evidence of harm whether people are born with very low cholesterol, if they maintain low levels naturally throughout life, or if they achieve very low levels with medical treatment. This is important: lower levels of blood cholesterol reduce disease risk but have no indications of directly causing any other problems. On the other hand, deliberately increasing blood cholesterol levels does not confer any direct health benefits. Some will cite studies reporting better health outcomes for people with higher cholesterol levels; this claim will be addressed in detail in section 3
Finally, the question implies an appeal to nature fallacy. That is, if something is natural, it must be good – or at least not harmful. Consider that our body also makes glucose (blood sugar), mainly in the liver. This glucose can similarly have harmful effects when it circulates through the blood at too high a level for too long. We see this in diabetes when glucose is overproduced by the liver and is not appropriately taken up by the tissues — much like blood cholesterol levels. The common claim that “our body wouldn’t make something that’s toxic!” reflects seemingly common sensical, but flawed reasoning. Our bodies do not always act in our own best interests.
Bottom line: Our cells are equipped to make essentially all the cholesterol they need without a large need for delivery of cholesterol from the bloodstream. Blood cholesterol tests do not tell us how much cholesterol is inside cells, where it is made and used for basic functions. Consistent evidence shows lower disease risk whether people are born with genetically low blood cholesterol levels, if they maintain naturally low blood levels throughout life, or if we deliberately lower blood levels with medications. This lower disease risk is apparent without evidence of harms — even with extremely low blood LDL-cholesterol. Conversely, no evidence shows an improvement in health by deliberately increasing blood cholesterol levels, for example by consuming a diet high in animal-derived saturated fats (assuming the person is not malnourished to start).
2 It’s not the cholesterol, it’s the inflammation
Inflammation is the body’s normal response to tissue damage, potentially harmful substances, or pathogens like bacteria. Inflammation is normally a controlled, temporary process. However, sometimes this inflammation can become uncontrolled and last too long. This can be severe in autoimmune diseases like Rheumatoid Arthritis and Lupus, or it can be milder in other conditions like Type 2 Diabetes and Chronic Kidney Disease.
Some argue that inflammation is what causes cardiovascular disease, not blood lipoproteins and cholesterol. It is true that long-term inflammation increases heart disease risk. This likely occurs in multiple ways, but long-term inflammation can affect our blood vessels and make them more prone to plaque development. In fact, certain anti-inflammatory medications such as Colchicine and Canakinumab have been shown to reduce the risk of heart attacks. Nidorf 2020 Ridker 2017
Inflammation can lead to the modification of certain substances through a process called oxidation. Oxidized LDL particles are associated with higher heart disease risk, but this relationship also disappears when we account for the total number of lipoprotein particles in the blood via apolipoprotein B measurement. It turns out that fully oxidized LDL particles do not circulate in significant concentrations in the blood, as these are rapidly cleared from the blood by the liver. Holvoet 2017 It is possible for apolipoprotein B-containing lipoprotein particles to carry oxidized phospholipid molecules, which do have harmful effects and appear to increase heart disease and stroke risk. Ultimately, the total number of potentially harmful cholesterol-carrying lipoprotein particles (approximated by apoB) over the lifespan is the most consistent independent predictor of heart disease risk. Wu 2006
A blood test called C-Reactive Protein (CRP) is often used to evaluate the amount of ongoing inflammation in the body. When looking at cholesterol levels and CRP levels, we find that both have their own independent relationships with overall risk. People who have both high cholesterol levels and high inflammation have the highest risk of heart attack. But when inflammation levels are low (CRP below 2 mg/dL), if cholesterol levels remain elevated (even if only mildly elevated) there is still a higher risk of having a heart attack compared to having low inflammation and low blood cholesterol. Storey 2018 This illustrates that it is not just about inflammation alone, and that cholesterol does matter. In fact, people can still develop plaque and experience heart attacks early in life in the absence of any inflammation or other risk factors like diabetes, high blood pressure, or smoking. Friera 2017 This is especially accelerated in people who are born with very high cholesterol due to genetics, known as Familial Hypercholesterolemia, even without any other medical problems or risk factors.
To further support these findings, the Multi-Ethnic Study of Atherosclerosis (MESA) study evaluated numerous biomarkers of inflammation as they related to heart disease risk. These biomarkers included C-reactive protein, interleukin-6 (IL-6), fibrinogen, and low-density lipoprotein particle size (discussed in section 4). All these markers were associated with plaque progression, but their significance disappeared after adjusting for traditional risk factors like smoking, high blood pressure, and diabetes. Zeb 2021
We should not ignore inflammation. If someone has an inflammatory disease it should be treated, just like any other issue that can impact health and longevity. We also have a variety of interventions that can decrease inflammation and improve conditions like type 2 diabetes and high blood pressure, such as regular exercise, adequate amounts of high-quality sleep, and a healthy dietary pattern centered primarily around fruits, vegetables, legumes, and unsaturated fats from fish and plant sources. Medications used to lower blood cholesterol levels like statins and ezetimibe also decrease inflammation in addition to their lipid-lowering effects — which can help people achieve the lowest possible risk when used appropriately.
Bottom line: Inflammation can play a role in accelerating the development of plaque and heart disease. However, even without significant inflammation, elevated cholesterol levels still result in higher risk. This risk increases further with even higher cholesterol levels, and the longer these levels remain high throughout life. Numerous markers of inflammation and oxidation in the blood lose their predictive value for heart disease once we account for the total number of apolipoprotein B-containing particles that can deposit cholesterol into the walls of blood vessels.
3 This study found that people with higher cholesterol live longer than people with low cholesterol, so high cholesterol improves longevity
There are observational studies that find patients with low cholesterol have higher rates of death and other diseases like cancer when compared to patients with higher cholesterol levels. These findings are typically found in populations over age 60 and have led to claims of a “cholesterol paradox.” This causes confusion regarding blood cholesterol as it relates to long-term health. Individuals who are skeptical of the established research use these findings to cast doubt on the relationship between blood cholesterol and disease risk. These studies have limitations and flaws that we detail below. The conclusion that cholesterol improves longevity cannot be drawn based on these findings.
Most lipoproteins are made by the liver and carry cholesterol, triglycerides, and other lipids around the blood. After making their trip around the body they are removed from the blood, mostly through the liver. Several treatments lower blood cholesterol levels including statins, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, and ezetimibe, among others. Although these work through different pathways, they ultimately increase the rate that low-density lipoprotein (LDL) particles are cleared from the blood. Treatments that lower blood levels of these lipoproteins consistently show predictable decreases in heart disease risk.
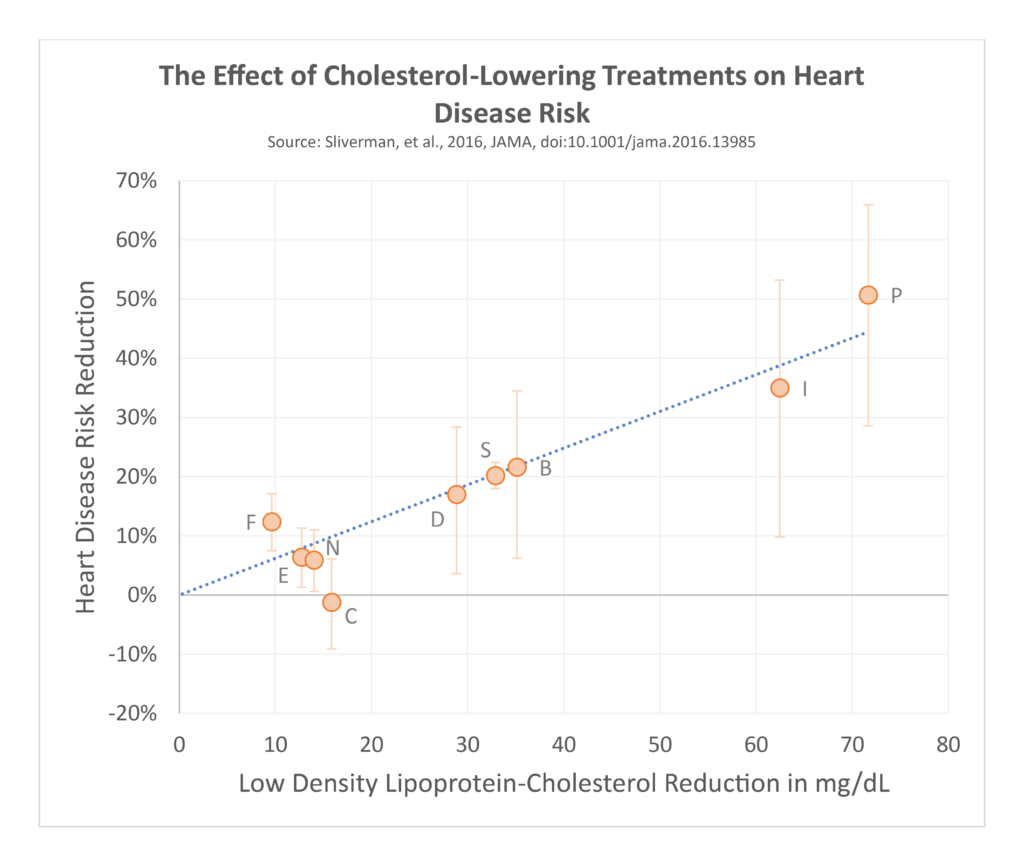
Clinical trials of these medications have included hundreds of thousands of patients, making them some of the most studied medications in existence. Collins 2016 This provides more than enough power to detect if we were directly causing major harms by lowering blood cholesterol levels. Across all randomized trials of various kinds of cholesterol-lowering medications, including those that treated participants to extremely low LDL-cholesterol levels, there has been absolutely zero effect found on the rates of cancer, and no increase in rates of death among groups treated with these medications versus control groups that received placebo treatment. CTTC 2019 Sabatine 2018 Although the massive amount of data from these randomized trials is already sufficient to dispel the myth that low cholesterol levels directly cause cancer, dementia, or death, we have other lines of evidence pointing in the same direction as well.
Genetic variation can have large effects on blood cholesterol levels from birth. People can have variations that result in severe elevations in blood cholesterol, or variations that result in much lower levels. Studies examining people with these different kinds of variations reveals striking differences. Those with genetically low blood cholesterol levels from birth tend to have the lowest incidence of heart disease, better long-term survival, and no increased risk of cancer. In contrast, those with the highest levels from birth have increased incidence of heart disease and worse survival. These relationships hold across the entire lifespan, including into the oldest age groups.
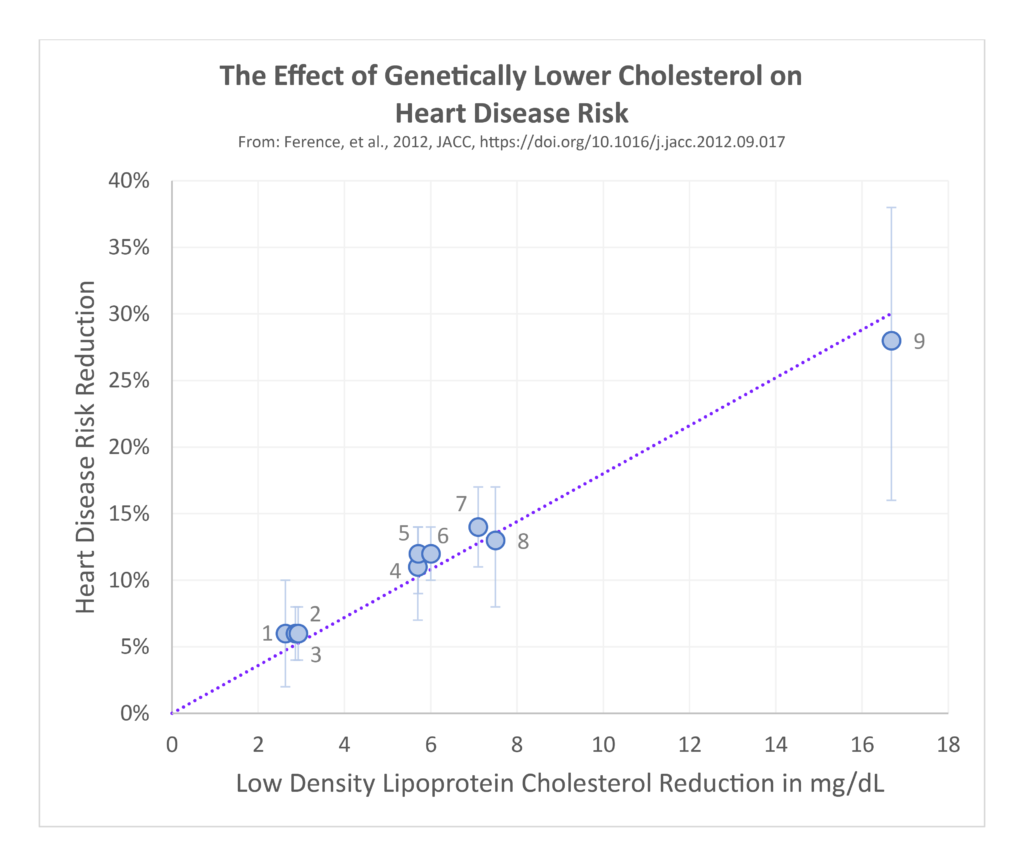
These genetic findings provide even stronger evidence to dispel the myth that higher cholesterol is protective or confers longevity. Postmus 2015 Similar observations are found in long-term prospective epidemiological studies, where people who start out with the lowest blood cholesterol levels in early middle age are followed upwards of 40–50 years later show better health outcomes compared to those starting with much higher blood levels. Hyttinen 2011
This brings up a major criticism of short-term or the studies finding lower risk of death among older individuals with high blood cholesterol. Specifically, they do not account for the overall effect of high cholesterol over the entire lifespan. These studies often observe patients all at one time (known as a “cross-sectional” study) or are very short-term, starting in later life. Those who had the highest levels starting earlier in life would have already died from cardiovascular complications, resulting in a type of “survivorship bias.” This is a logical error where researchers look only at the people who are living and available at the time of study, while ignoring those that have already died, resulting in flawed conclusions. For another good example of this, see this Wikipedia article on the topic. Fortunately, the genetic data discussed above reveal the true effect of lifelong exposure to high versus low blood cholesterol levels, and this is further supported by prospective studies that follow patients forward over time, with or without medical intervention.
Finally, in contrast to people who have genetically low cholesterol from birth, there are conditions that develop later in life that can result in a decrease in blood cholesterol levels for other reasons. Many chronic diseases can affect lipid metabolism and liver production of cholesterol and lipoproteins. Cancer, chronic liver disease, cachexia, and other more advanced forms of chronic organ failure can all cause these changes. As a result, people with these conditions often have lower blood cholesterol levels. However, they have a higher overall risk of death due to their underlying (and often undiagnosed) cancer or other chronic disease. This increased risk is not due to the low cholesterol itself.
If low cholesterol directly caused more disease or death, this would be apparent in genetic studies that follow people with low cholesterol from birth over the lifespan. No such finding has emerged. In reality we observe the opposite.
Bottom Line: Findings that people with higher cholesterol levels have a lower risk of death (or vice versa) are artifacts of flawed observational research. These studies fail to account for the effect of total lifelong exposure to blood cholesterol and are prone to survivorship bias. Certain advanced disease states can result in a natural lowering of cholesterol, which can also contribute to this type of finding. Genetic studies examining people who have low versus high blood cholesterol levels from birth find that lower levels confer better health and longevity without an increased risk of cancer, while high levels from birth increase the risk of premature disease and death. Randomized trials of medications used to lower blood cholesterol similarly find no increased risk of cancer or death, regardless of how low LDL-C becomes. These lines of evidence refute the myth that high cholesterol confers longevity.
4 Whether you have large or small LDL particles is more important than the number
Plaque development begins when lipoprotein particles penetrate the blood vessel wall and deposit their cholesterol, which causes a local inflammatory response. There are enormous numbers (quintillions) of lipoproteins circulating in our bloodstream with a variety of different sizes, contents, and characteristics. The different types of particles carry variable amounts of cholesterol and triglyceride, and this results in differences in the overall size and composition of the lipoprotein particle as it travels through the blood. Lipoproteins smaller than 70 nanometers (nm) in size are small enough to penetrate the blood vessel wall and initiate the process of plaque development. Tabas 2007
Some people claim that large, “fluffy” low-density lipoprotein (LDL) particles are less prone to causing plaque, whereas small, “dense” LDL particles are the real source of risk for plaque and heart disease risk. Specialized testing can indicate whether someone has mostly larger particles, known as “pattern A”, or mostly smaller particles, known as “pattern B”. Smaller, pattern B particles are more common in people with insulin resistance and diabetes. Let’s take two hypothetical patients and their blood cholesterol tests:
Patient 1:
Low-density Lipoprotein Cholesterol (LDL-C): 140 mg/dL, pattern A (larger particles)
Patient 2:
Low-density Lipoprotein Cholesterol (LDL-C): 140 mg/dL, pattern B (smaller particles)
In this scenario, Patient 2 is indeed at higher risk than Patient 1. The question is, “Why?” Is it actually because their particles are of smaller size? As it turns out, no.
Patient 1 has larger LDL particles than patient 2, but both are carrying the same total amount of LDL-cholesterol, 140 mg/dL. Because patient 2 has smaller particles, they need a higher total number of LDL particles to carry this same amount of cholesterol. Remember that 140 mg/dL is the mass of low-density lipoprotein contained in a volume of blood. Smaller particles carry a lower mass of cholesterol. Therefore, you need more pattern B particles in a given volume of blood to get to 140 mg/dL than you would with pattern A particles. The higher total number of particles in the blood increases the chance that they will get into the blood vessel wall.
Large, pattern A LDL particles are defined as those greater than 25 nanometers in size (average about 27 nm), whereas small, pattern B particles are defined as those less than 25 nm in size (as small as 18 nm). Williams 1992 This means that both large and small particles are still well below the 70-nanometer threshold that allows particles to penetrate blood vessel walls.
A variety of experiments have measured blood cholesterol levels, particle sizes, and compared these with measurements of total LDL particle numbers. Apolipoprotein B measurements allow particle numbers to accurately analyzed. Patients with small LDLs do have higher risk than those with larger LDLs if we do not account for overall particle number. However, this apparent effect of size disappears when we account for total particle number. Ip 2009 Mora 2007 When you have the same number of LDL particles, regardless of whether they are pattern A or B, risk is essentially the same.
This has some interesting implications that have been confirmed in the research as well: for someone with high LDL-C but with low apoB, indicating a small number of particles carrying plenty of cholesterol, heart disease risk is low. Conversely, for someone with low LDL-C but high apoB, indicating a high number of particles carrying relatively little cholesterol, heart disease risk is high. Pencina 2015, Wilkins 2016, Lawler 2017
This illustrates how, from the standpoint of heart disease risk, “a particle is a particle,” and risk principally tracks with apoB levels. Marston 2021 Particle size does not influence this risk outside of its effect on total particle number. This should make sense since both large and small LDL particles are still plenty small enough to penetrate blood vessel walls. It is therefore not necessary or helpful to perform lab tests checking particle size, but is why we recommend measuring blood apolipoprotein B levels. Focusing on lowering this particle number will provide the main benefit. If a person has insulin resistance or diabetes (and is therefore prone to having smaller, and more numerous particles), addressing those issues through lifestyle and medical treatment would provide additional benefit for heart disease risk.
Bottom Line: While patients with predominantly small, dense LDL particles do appear to be at higher risk for cardiovascular disease, when the total number of particles is accounted for, this apparent “size” effect disappears. Total particle number is therefore the independent predictor of risk, without a significant effect of large versus small LDL particles on their own. Whether large or small, all LDL particles are still small enough (less than 70 nanometers in size) to penetrate the vascular wall and initiate the process of atherosclerosis. In practice, measuring Apolipoprotein B provides a metric of particle number — and is thus a superior tool for decision making compared to total cholesterol or LDL-cholesterol.
5 This study shows statins don’t work in the elderly
The risk of heart disease is strongly related to the cumulative exposure to cholesterol-carrying lipoproteins in the blood over the entire lifespan. While other risk factors such as chronic inflammation, diabetes, or smoking can accelerate this process and deserve treatment (see section 2), blood lipoprotein levels are central, since they can still cause plaque development even if those other factors are absent. This means that the earlier an individual develops high blood cholesterol, and the longer this elevation is maintained, the higher an individual’s overall lifetime risk of heart disease. Robinson 2018 Conversely, the earlier in life we can lower these blood levels, and the longer we can keep them low, the bigger of an impact we can have on the person’s overall lifetime risk. We have multiple lines of supporting evidence for this understanding.
By the time an individual is elderly, most of the cumulative lifelong exposure has already happened. The damage has already been done. It is therefore unsurprising that starting cholesterol-lowering treatment in advanced age has a much more limited ability to lower a person’s risk. It takes a much larger reduction in blood cholesterol in older age to achieve the same decrease in risk as a smaller reduction earlier in life that is maintained over the long-term. PSC 2007
For elderly patients at very high risk, for example those who have already had a heart attack, these medications still provide benefit. For elderly patients who are taking these medications, observational studies have found that stopping them is associated with an increase in risk — although there are limitations to these studies given their observational nature. Giral 2019 In some situations, this tradeoff in risk for a very elderly person is acceptable in order to reduce the complexity of their medication regimen (known as “pill burden”) or if their life expectancy is very limited for another reason.
Bttom line: heart disease risk is related to the cumulative, life-long exposure to lipoproteins and cholesterol in the blood. Interventions to lower blood lipoprotein and cholesterol levels early in life provide the greatest long-term decrease in risk. The longer intervention is delayed, the more cholesterol needs to be lowered to achieve the same benefit. By advanced age, most lifelong exposure has already occurred, and it becomes increasingly difficult to impact risk by lowering blood cholesterol at that point. The claim that these treatments have much smaller benefits in older age fits completely with our current understanding of how cardiovascular disease develops.
6 This study found that lots of patients who have heart attacks had normal cholesterol levels
Another common objection to the claim that blood cholesterol plays an important role in heart disease risk is the observation that a substantial fraction of individuals who experience a heart attack are found to have “normal” cholesterol levels (total cholesterol less than 200 mg/dL). At first, this may seem to be a compelling argument. Let’s take a closer look.
We have known for decades, dating as far back as the 1960s Framingham study, that about a third of heart disease occurred in people with levels of total cholesterol within the apparently “normal” range. Kannel 1961 Castelli 1988 Heart disease risk quadrupled in people with total cholesterol over 260 mg/dL, but the finding in apparently normal patients was still notable. Multiple studies from the 1960s through the 1980s similarly showed rapidly increasing heart disease risk as blood cholesterol levels increased well into these ranges. Martin 1986 These findings led to the first official clinical guidelines from the Adult Treatment Panel I, which established a cutoff of 200 mg/dL or less as “desirable blood cholesterol levels”. Goodman 1988
It is important to understand that “normal ranges” are, to an extent, arbitrary. For example, “Recommended Dietary Allowances” (RDAs) in nutrition are basically the amount of a given nutrient that would be necessary to meet the requirements of 97% of healthy people. Why 97%? It needed to be set somewhere and 97% captures a lot of the population. Still, there is nothing magical about 97%. The RDAs would change if the cutoff had been arbitrarily set at 90% or at 99.5%.
Similar issues arise elsewhere in medicine. Diagnostic cutoffs for “high blood pressure” have decreased over the years as more evidence has emerged to help us understand thresholds and spectrums of risk. Saklayen 2016 The same original Adult Treatment Panel I recommendations from 1988 recognized these issues, stating (emphasis ours):
“Because the relationship between serum cholesterol level and CHD is a continuous and steadily increasing one (Fig 1), these cut-points are necessarily somewhat arbitrary. However, this is also true of other risk factors, such as blood pressure, and the success of basing clinical decisions on whether or not a patient is classified as hypertensive indicates the value of establishing cut-points for clinical decisions. The 240 mg/dL cut-point for total serum cholesterol is a level at which CHD risk is almost double that at 200 mg/dL, and is rising steeply.”
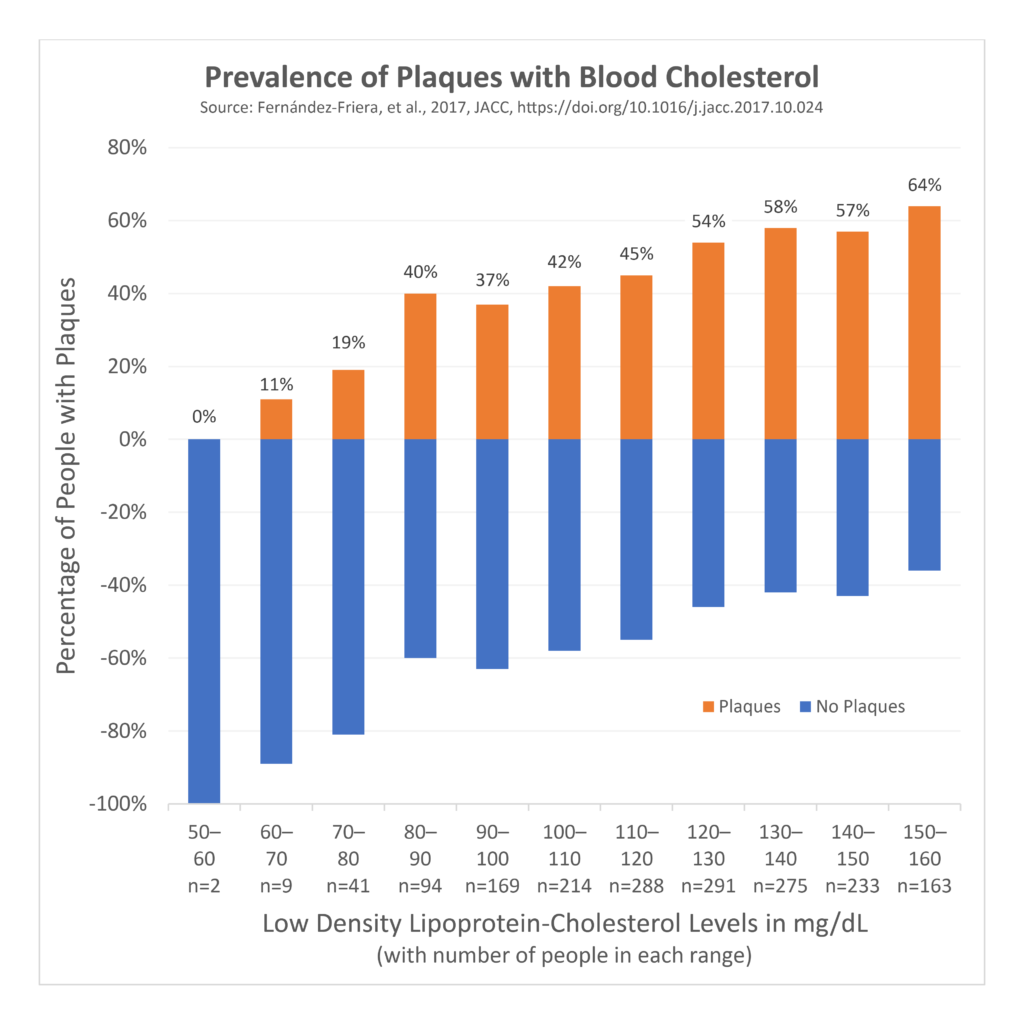
Since that era, we have learned much more about the relationship between blood cholesterol levels and the risk of developing plaque and heart disease. A 2017 study looked at 1,779 people who did not have any conventional risk factors for heart disease, like smoking, high blood pressure, diabetes, and very high cholesterol (according to “normal” ranges). Friera 2017
Despite having no other conventional risk factors, half of these people were still found to have plaque in their arteries, and blood LDL-cholesterol levels were linearly and independently associated with the amount of plaque. Plaque was found in 11% of those with LDL-cholesterol of 60–70 mg/dL, and this linearly increased up to 64% of those with LDL-cholesterol of 150–160 mg/dL. Remember that the normal level is set at 200 mg/dL and a few people with 60–70 mg/dL had evidence of plaque. Even with LDL-cholesterol levels that were within the supposedly “normal” range, AND without other conventional risk factors for heart disease, the more LDL-cholesterol, the higher the risk of plaque development. We also know from other evidence that lowering blood LDL-cholesterol to below 70 mg/dL halts plaque progression. Ahmadi 2019 These findings should raise questions about our “normal” range and what criteria we should use to set it. Even the 1960s Framingham study authors wrote (emphasis ours):
“In evaluating the risk associated with elevation of serum cholesterol levels it is important to recognize that in the Framingham population the reference base for serum cholesterol (i.e., “normal” cholesterol) may be high when compared with some other populations in which lower rates of [heart disease] have been claimed.”
Similarly, in the 2017 paper described above, the authors wrote (emphasis ours):
“Importantly, LDL-C levels in our population were well within the range considered normal, reinforcing the concept that desirable LDL concentrations are probably much lower than those currently recommended.”
That is important. The cutoff of 200 mg/dL may be set too high. Based on this evidence, and since all cut-offs are to some extent arbitrary, we should likely reconsider what we think of as “normal” in order to provide better descriptions of risk for making decisions with patients. This is what has already been done with blood pressure cut-offs as we recognized the spectrum of risk even at levels below what was previously considered “high”, and the same likely needs to happen with blood cholesterol cut-offs.
When someone says that a significant number of patients experience heart attacks with “normal” cholesterol levels, they are correct — this is simply a reflection of the fact that our “normal” cut-offs are not low enough to sufficiently distinguish people who are truly at lower versus higher risk of disease. It is problematic to dichotomize a large spectrum of risk into two neat, separate categories of “normal” versus “high”. It is also true that heart attacks have more than one cause. Having high blood cholesterol over a long period of time is one of those causes.
There are two additional considerations in this topic. First, certain individuals may demonstrate a phenomenon known as “discordance”, whereby blood LDL-cholesterol levels may appear normal, but they have much higher numbers of particles to carry this cholesterol. This was discussed in section 4 above, on “large” vs. “small” particle sizes. This commonly happens in people with insulin resistance and diabetes. In these situations, particle number (as measured by Apolipoprotein B) tells the true story in terms of risk, rather than the LDL-cholesterol. Marston 2021 Given that many people do not get ApoB checked, but exclusively rely on LDL-cholesterol, having undetected discordance can result in a false reassurance about risk.
Finally, there are a variety of changes in the blood that happen in the context of physiologic stressors like heart attacks, strokes, infections, and inflammation. This is due to a process known as the “acute phase response”. Certain substances known as “acute phase reactants” increase during the acute phase response, including C-reactive protein, Ferritin, and the Erythrocyte Sedimentation Rate, among others. Other blood concentrations decrease in the acute phase response; these substances are known as “negative acute phase reactants”. These include things like albumin, vitamin D, testosterone, and, most relevant for this context, blood low-density lipoprotein and total cholesterol levels. Balci 2011 This means that if someone experiences a medical issue (for example, a heart attack) and one of these negatie acute phase reactant tests are checked, like cholesterol, low-density lipoprotein, testosterone, or vitamin D, the results will be falsely low. In other words, upon hospitalization for something like a heart attack, blood LDL-cholesterol levels will be low due to the acute phase response. This test does not tell us anything about the lifetime of blood cholesterol levels the person had experienced leading up to that point. This means that studies finding strong associations appear between how low these levels get, and how sick someone becomes (or their risk of death) will result in erroneous conclusions.
Heart disease risk is related to how high blood cholesterol levels are elevated over the course of the entire lifespan – and a single blood test, perhaps measured at the time of hospital admission for a heart attack, tells you nothing about this lifetime exposure.
Bottom line: All diagnostic cutoffs in medicine are, to some extent, arbitrary. The “normal” range for total cholesterol (less than 200 mg/dL) was set in the 1980s based on evidence that heart disease risk started rapidly increasing as total cholesterol levels increased beyond this level. But even at that time we already had ample evidence that a third of heart attacks happened in people who had levels below this cutoff. From more recent evidence we know that even among people with no other risk factors for heart disease, LDL-cholesterol has a linear, independent relationship with plaque development throughout the normal range, increasing as levels go up beyond 70 mg/dL. We also know that lowering LDL-cholesterol below 70 mg/dL halts plaque progression. In the same way diagnostic cutoffs for high blood pressure have been lowered over time as we learned more about the spectrum of risk, we should likely do the same with diagnostic cutoffs for “normal” cholesterol levels. Apolipoprotein B levels provide a superior estimate of risk, particularly if patients are at risk for discordance with LDL-cholesterol levels due to insulin resistance or diabetes. Finally, we must recognize that blood lipoprotein levels can change during a physiologic stressor like a heart attack or infection, and observational studies that check these levels during such an event produce unreliable associations and conclusions.
7 If your Coronary Artery Calcium score is zero there’s nothing to worry about
Plaque development begins when lipoprotein particles penetrate the blood vessel wall and deposit their cholesterol, which causes inflammation in the area. As the disease progresses over time, calcium can become deposited around plaques in the arteries. Mori 2018 This calcium lights up on imaging tests like CT scans, which has led to special tests to look for “Coronary Artery Calcium”, or CAC. A CAC “Score” is generated to reflect how much calcium is present in blood vessel walls. It is measured in terms of Agatston units, as follows:
| Coronary Artery Calcium Score (Agatston Units) | Heart Disease Severity |
| 0 | None |
| 1–99 | Mild |
| 100–399 | Moderate |
| 400 or greater | Severe |
Individuals with a CAC score of zero have a much lower risk of having a heart disease complication like a heart attack over the short- to medium-term compared to people with higher scores. Hussein 2020 Many have used this evidence to claim that if their CAC score is zero, that they have no plaque and are therefore at zero risk. This is sometimes used to justify ignoring (or not treating) risk factors like elevated blood cholesterol.
This approach can be shortsighted. Calcium deposition in a plaque happens later in the disease process and is therefore a marker of more advanced disease. It is rare to find calcium in the arteries of young people, whereas it is still possible for them to have “soft” plaques that do not appear on a coronary artery calcium scan. Gabriel 2018 Kim 2012 In fact, about one-third to one-fourth of heart disease complications in young people occur in people with a CAC score of zero. Interpreting someone’s lifelong risk of heart disease is more complex than a CAC score alone.
The meaning of calcium can also vary based on the context; for example, high-intensity exercise and statin medications are also known to modestly increase coronary calcium without increasing heart disease risk. DeFina 2019 In fact, both exercise and statin medications reduce the risk of heart disease complications despite causing mild increases in coronary artery calcium. CTTC 2019 It is thought that these interventions alter plaque composition, and that this calcium may have a “stabilizing” effect on the plaque that makes it less likely to rupture and precipitate a heart attack or other complication. Puri 2014 Puri 2015
A CAC score of zero is very reassuring in an older person where we’d typically expect to find lots of calcium but is much less useful in young people. Similarly, finding calcium in a healthy endurance athlete without other risk factors requires careful interpretation. Plaque development and progression relates to the total lifelong elevation in blood cholesterol — so even for a person with a CAC score of zero right now, lowering these blood levels using all the strategies discussed in part 2 is still recommended, especially if they have many years left to live.
We do not recommend that all people obtain CAC screening tests. There are even arguments against checking CAC at all in many situations. Mandrola 2019 We recommend an individualized approach in collaboration with a physician that includes measuring BMI and waist circumference, blood pressure, blood cholesterol and apolipoprotein B levels, and potentially testing for diabetes or other risk factors depending on the individual situation. Additionally, if a CAC score is obtained and is zero, consider repeating it in 5 to 10 years to assess for changes. Lehman 2018 If the initial score is greater than zero, repeating the score over time is less likely to be helpful; instead, we recommend more aggressively addressing individual risk factors like physical activity, body fat, blood pressure, apolipoprotein B levels, and other individual issues.
Bottom Line: Calcium deposition into plaques tends to occur in later stages of disease. It is still possible to have plaque without calcium. Conversely, it is also possible for calcium to deposit for other reasons without an increase in risk, or even despite decreases in risk, as is observed with long-term high-intensity exercise or with the use of statin medications. This makes CAC interpretation complicated. A CAC score of zero does not mean that a person has zero risk of heart disease complications, or that they have zero plaque in their blood vessels. A CAC score of zero provides the most reassurance for an older person where we’d otherwise expect to find calcium, whereas it provides less reassurance among younger people and women, where it is expected to find none. Even for people who have a CAC score of zero, we should still emphasize the management of risk factors like obesity, blood pressure, blood cholesterol, and others, especially if they have many years of life expectancy left.
8 Statins cause lots of problems and you’ll lose all your gains
Statins are commonly used medications to lower blood cholesterol levels and the risk of heart disease complications. These compounds were discovered in the 1970s and underwent clinical studies before approval for use in 1987. Since then, simvastatin, pravastatin, atorvastatin, rosuvastatin, and several others have become available for clinical use. Regardless of which how it’s done, lowering blood LDL-cholesterol by 1 mmol/L (about 38 mg/dL) consistently results in a 22–24% decrease in the risk of heart disease complications. Ference 2017 Statins fit right in with these data alongside medications like ezetimibe, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, or other medications, or if the LDL-cholesterol is lowered through diet.
While there are subtle differences among the different statins, all work by blocking an enzyme our bodies use to create cholesterol called HMG-CoA Reductase. This causes cells to increase how many LDL receptors they express on their outer surface, to “suck up” more LDL particles and their associated cholesterol out of the blood. As a result, blood cholesterol levels fall as it gets pulled into the tissues. This accelerated clearance from the blood means lipoprotein particles have less opportunity to penetrate the blood vessel wall and drive the development and progression of plaque.
Despite these established benefits, side effects are a very common concern. Statins, like every other medical intervention, offers potential benefits as well as potential risks that must be weighed in the context of an individual person’s situation. The potential benefits include a decrease in the risk of plaque development and complications from cardiovascular disease, as mentioned above.
One common concern is muscle-related symptoms from statin use, including muscle aches (known as myalgia), or muscle damage (known as myopathy). These concerns are especially prevalent among lifters and other strength athletes concerned with their muscles. Significant muscle damage is extremely rare, occurring in less than a fraction of one percent of users. It depends on the specific medication and dose being used, as well as certain patient factors. A much larger proportion of patients report muscle aches without objective evidence of muscle damage on blood tests. Let’s look at this last concern more closely.
In studies comparing statins to placebo pills, when patients know which option they are taking, side effects are reported more frequently with statins. However, when they are blinded to which option they’re taking (that is, they are unaware if they are taking a statin or placebo), these muscle-related side effects are reported at the exact same frequency. In other words, when taking a fake pill, people reported identical levels of muscle aches as people taking statins. This has been shown in numerous studies, including one randomized trial of over 10,000 patients. Gupta 2017 This finding was investigated further in the 2020 SAMSON trial, where 60 patients who had previously stopped statins due to perceived side effects were given four pill bottles (one statin, two placebo, one empty) and randomized to take them in different orders. Wood 2020 They rated side effect intensity on a scale of 0 to 100 every day. By the end of the study, there was no statistical difference between ratings of side effects while taking placebo pills or statins, but these were both about double the ratings of side effects during “no-pill” months.
In other words, the patients’ ratings of side effects were the same whether they were taking a real statin or a placebo pill, and both were significantly higher than when taking no pill at all. Researchers found that 90% of the symptoms reported by patients taking a statin were also reported when they took a placebo pill. Overall, the on this topic shows that the muscle aches are not clearly due to the drug itself, but rather due to the expectation or concern about such side effects just from taking a pill. This expectation of negative effects is known as a “nocebo” effect.
While there are several theoretical mechanisms by which statins could have other negative effects on muscle function, when these medicines have been studied in humans, we do not find that the medicines consistently decrease muscle strength, aerobic capacity, or other objective metrics of muscle function. Noyes 2017 Studies examining subjective metrics around exercise are limited by the nocebo issue described above, since patients are often not blinded to whether they are taking a statin. In the rare situations of more severe adverse effects mentioned above, discontinuing the medication would be necessary and treatment with alternative options considered. Otherwise, for an individual with higher risk of heart disease, the benefits of these treatments alongside lifestyle modifications outweighs most of the theoretical risks that have not been borne out in controlled trials in humans. In fact, for people at very high risk due to genetically high cholesterol from birth, we have data over 20 years of follow up while taking statins showing their benefit in reducing the risk of heart attacks and death. Luirink 2019
Other common concerns around statin medicines relate to memory loss and cancer risk. Fortunately, we have lots of data from randomized, controlled trials on these questions as well. Regarding memory loss and dementia risk, one trial of more than 20,000 patients randomized to statin versus placebo found no difference in the development of cognitive impairment or dementia over the course of 5 years, including among the subgroup of patients aged 75-85 years. HPSCG 2002 The very large sample size of this study provides the power needed to detect such differences over the shorter time span of 5 years. Another randomized trial of 5,804 patients randomized to statin versus placebo similarly found no difference in cognition over the follow up period. Trompet 2010 Even studies examining the use of statins in patients who already have Alzheimer’s disease have shown no effect on progression of disease. Sano 2011
Regarding cancer risk, data from prospective randomized, controlled trials has shown no effect whatsoever on the risk of cancer at all – neither benefit nor harm. This may contrast with the findings of certain retrospective or other observational studies, but the evidence from prospective, randomized studies supersedes observational data in this context. Collins 2016
For patients who are at high risk of heart disease complications but have difficulty tolerating a statin medication, there are several options that may be beneficial. Switching to a different statin, trialing a lower dose (potentially in combination with a non-statin medication, such as ezetimibe), taking the medication at bedtime instead of in the morning, or trialing every-other-day dosing, particularly of the longer-acting statins, may be feasible options to lower risk while managing perceived side effects.
Bottom Line: Statins reliably lower blood lipoprotein and cholesterol levels, as well as reducing the risk of heart disease complications — with greater benefits the earlier in life that blood lipid levels are brought under control. While concerns about muscle-related side effects are common, at least 90% of these effects are reported when patients are blinded to whether they are taking statin or placebo, regardless of which pill they are taking. This suggests that these reported effects are mostly attributable to a nocebo-type effect and are not due to unique drug effects. Finally, we do not have evidence that these medicines reliably lower objective metrics of muscular adaptation such as strength, hypertrophy, or aerobic capacity. True muscular injury from these medicines is exceptionally rare, occurring in less than a fraction of a percent of users, which is typically outweighed for most patients by much higher risks of heart disease-related complications. Evidence from prospective randomized, placebo-controlled trials do not show increases in the risk of cancer, cognitive impairment, or dementia from the use of these medicines. For those who are intolerant of a statin, there are several options for altering the regimen to improve tolerance.
9 This study found no relationship between high vs. low saturated fat intake and heart disease
Hundreds of controlled feeding studies as well as other lines of evidence over the past century have shown that animal-derived saturated fat intake tends to increase blood levels of low-density lipoproteins (LDL) and the cholesterol they carry. Clarke 1997 We also have decades of mechanistic, genetic, epidemiological, and interventional studies showing that the lifelong exposure to low-density lipoproteins in the blood is the primary causal driver of plaque development and heart disease risk. Ference 2017 Boren 2020 However, numerous studies report a lack of relationship between dietary saturated fat intake and the risk of heart disease. This seems paradoxical. These papers seem to cast doubt on the ideas presented throughout this series, but this area of research can be extremely tricky to interpret. In this section we will discuss the research issues that lead to these erroneous conclusions.
Animal-derived saturated fats relate to plaque development and heart disease risk indirectly, by altering blood lipoprotein and cholesterol levels as an intermediate step. The common idea of dietary fat directly “clogging” arteries and precipitating heart attacks is not accurate. Rather, the causal sequence involves dietary saturated fats (and other factors) increasing blood lipid levels, which then drive plaque development and progression over time.
We know that there is a threshold effect of dietary effects on blood cholesterol levels, particularly as intake rises from low levels (less than 10% of total calories) to high levels (greater than 18-20% of total calories). Conversely, we observe decreases in disease risk when this intake is reduced from those high levels to less than 10% of total calories, depending on what foods are substituted. Hooper 2020 Li 2015
- Replacement with polyunsaturated fats (such as from fish, nuts, and other plant sources) results in a large decrease in blood cholesterol and risk.
- Replacement with plant-derived monounsaturated fats (nuts, olive oil, avocado), or with fibrous carbohydrates (legumes, oats) results in a moderate decrease in blood cholesterol and risk. Animal-derived monounsaturated fats tend to be found in meats and increase risk.
- Replacement with refined carbohydrates like sugar has no significant effect, which suggests that these are essentially equal in terms of their effect on blood cholesterol and risk.
- Replacement with trans-fats increases risk, which is why these have been banned from the food supply – although this was not the case when several older studies on this the topic of saturated fat replacement and heart disease risk were performed.
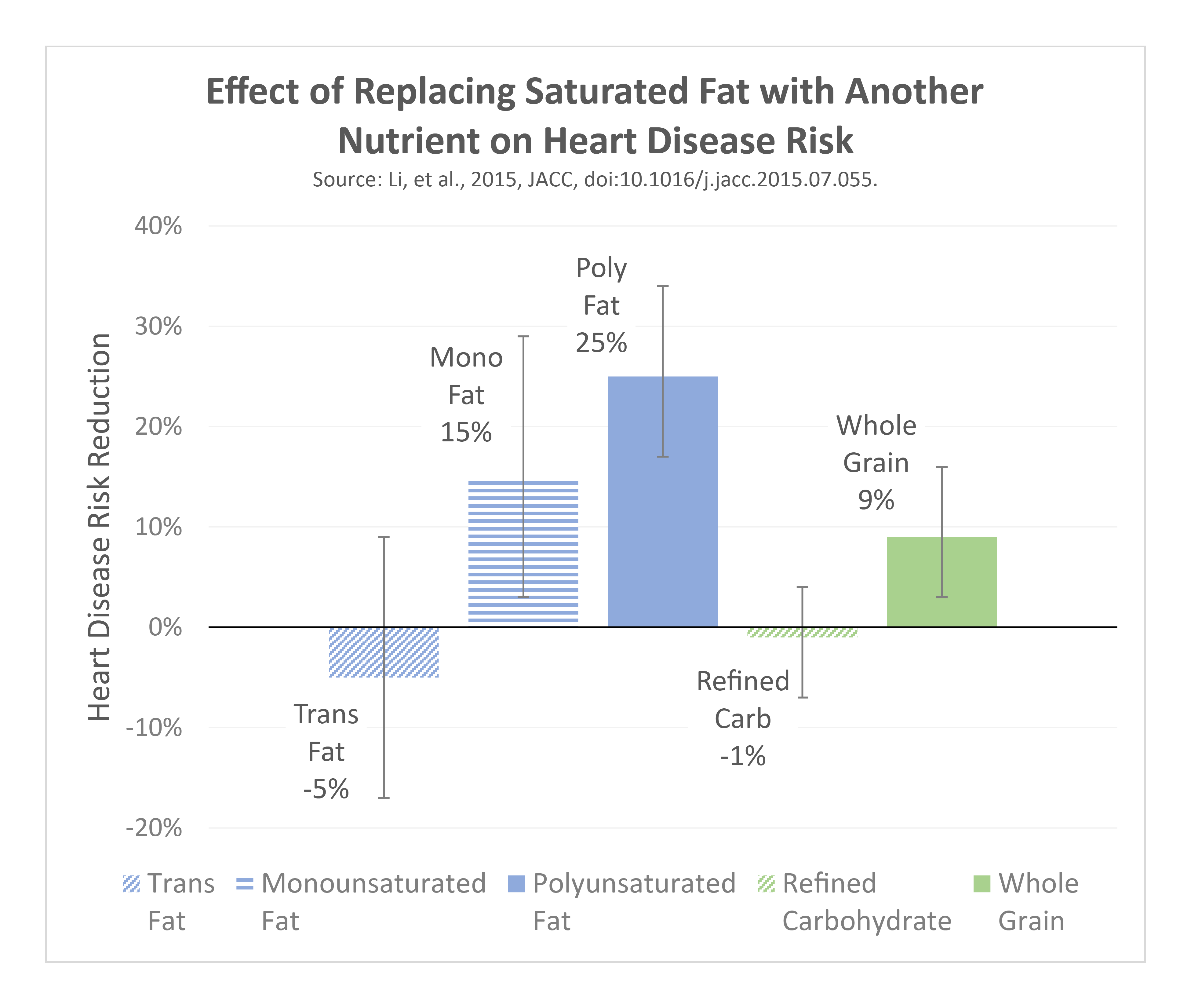
Saturated fats from dairy products (not including butter) do not appear to confer the same increase in risk as compared with saturated fats from butter and fatty animal meats. Butter is made by separating cream from milk, then separating the remaining fat through churning. This process results in a final product that is much higher in fat, lower in calcium, and that lacks a protective compound known as milk fat globule membrane (which is preserved in milk and cheese). The removal of this compound appears to contribute to the harmful effects of butter compared to other forms of dairy like yogurt. Rosqvist 2015 As a result, these other dairy fats should not be “lumped in” with all saturated fats.
These nuances become relevant when analyzing research studies on this general topic. Studies examining the relationship between saturated fat intake and heart disease risk must specifically account for several factors:
- How much is “high” versus “low” intake? Is there sufficient difference between groups for a study to cross the threshold of effect? To call a diet high in saturated fat, we are looking for intakes greater than 18–20% of calories. Conversely, diet low in saturated fat should have intakes below 10% of calories. Comparing these sufficiently different intake levels would provide the clearest contrast across the known threshold of effect. In contrast, a study that compares a “high” intake of 18% of calories from saturated fats versus a “low” intake of 12% will be less likely to detect a difference in outcomes due to insufficient contrast between groups. Similarly, comparing 10% versus 4% does not cross the threshold either. When studies claiming to compare “high” versus “low” actually look at “high vs. high”, or “low vs. low”, we are set up to find no differences and draw erroneous conclusions.
- What are the food sources of saturated fats? For example, is it primarily from dairy products that pose little to no risk, or other foods that markedly increase risk like butter and fatty animal meats? What foods replaced these saturated fats to take a truly “high” intake down to truly “low”? We expect decreases in risk when replacing with polyunsaturated fats, plant-derived monounsaturated fats, or complex carbohydrates. We expect no change or an increase in risk from refined carbohydrates, animal-derived monounsaturated fats, or trans-fats.
- How did the study account for the indirect effect of saturated fats on heart disease risk, via the intermediate step of affecting blood levels of LDL? If a study controls for blood cholesterol levels between groups consuming high versus low saturated fat diets, it is essentially erasing the intermediate step of the causal sequence. This would erroneously bias a study to find no difference when there truly is a difference present.
With these basics in mind, we can observe examples of common flaws in studies that claim a lack of relationship between “high” versus “low” dietary saturated fat intake, and the risk of heart disease.
9.1 Contrast
The first common flaw involves inadequate contrast between “high” (greater than 18–20% of calories) and “low” (less than 10% of calories) intakes of saturated fat among study groups. The Prospective Urban Rural Epidemiology (PURE) investigation conducted from 2003 to 2013 was a large cohort study across 18 different countries and reported findings that higher saturated fat intake was associated with a lower risk of stroke. Dehghan 2017 Dietary saturated fat intake was divided into quintiles (fifths); in Asian regions, four of the five quintiles consumed between 2% and 7%, with the top quintile still only consuming 12% of calories from saturated fat. In non-Asian regions, the bottom three quintiles were between 5% and 9%, with the top two quintiles at 11% and 14% respectively. These make it difficult to test “high versus low” as defined above, particularly when combining variable dietary patterns across 18 different countries. One of the study’s frequently-cited findings was that there is a higher risk of stroke observed at very low intakes of saturated fats. This likely reflects that in many of the included countries, the diet with 2% saturated fat is nutritionally inadequate in general. This finding does not allow us to conclude that consuming very high amounts of saturated fats will reduce our risk of stroke, nor does it directly test the impacts of consuming greater than 18–20% of calories from saturated fats versus 10% or less.
Similarly, the Malmo Diet & Cancer study from 1991 to 1996 compared a bottom quartile consuming 13% of calories from saturated fat against an upper quartile consuming 22%. The authors themselves wrote, “note that only 1.2 percent of the present study population actually followed national Swedish recommendations (less than 10 energy percent) on saturated fat intake. Strictly speaking, the SFA-CVD hypothesis is thus not fully testable in this population”, essentially recognizing that their study cannot draw conclusions on the question at hand.
There are many other trials that fail to account for this true “high versus low” comparison, limiting their ability to draw useful conclusions on the topic. This is a critically important feature to examine closely anytime someone cites a study that claims no difference across different levels of intake.
9.2 Food Sources & Replacement Effects
The next common problem can arise when studies fail to account for variation in the effects of food sources when replacing saturated fats with other nutrients. Some are beneficial, such as polyunsaturated fats and fibrous complex carbohydrates, whereas others are equally harmful or even more harmful, such as animal-derived monounsaturated fats, refined carbohydrates, or trans-fats. We also know that saturated fats from dairy sources (excluding butter) do not seem to confer the same degree of risk as foods like butter and fatty meats.
The 1960s Sydney Diet-Heart Study reported an increased risk of heart disease complications and death when saturated fats were substituted with polyunsaturated fats – which would seem to contradict what we have described thus far. However, the polyunsaturated fats in this study came from margarine, which during the 1960s contained high amounts of trans-fats (the kind which have since been banned from the food supply). This is an obvious source of error when drawing conclusions about the effects of this replacement nutrient, since it is well-established that trans-fats dramatically increase heart disease risk and should not be consumed.
The PURE study mentioned above reported higher mortality with higher carbohydrate intake but failed to differentiate between refined carbohydrates and complex carbohydrates. These are well-known to have divergent effects as replacement nutrients in humans, and accounting for these differences is critical to drawing accurate conclusions.
The Malmo study referenced above compared groups consuming 13% of calories from saturated fat against an upper quartile consuming 22%, but the authors also note “Foods contain many nutrients and other bioactive substances that interact in complex ways and may therefore differ in their health effects in ways not captured by differences in the content of single nutrients.” This is important since they recognize that dairy products are an important source of saturated fats in the Swedish population, but are unable to fully account for this in their study
A large meta-analysis by de Souza in 2015 concluded that, “Saturated fats are not associated with all-cause mortality, CVD, CHD, ischemic stroke, or type 2 diabetes”. de Souza 2015 That is a strong, declarative statement. However, when looking more closely at the study methods, they add:
“Most studies in the present review did not explicitly model the effects of nutrient substitution … Indeed, carbohydrate energy was typically lowest in those in the highest intakes of saturated and trans-fat. Common sources of carbohydrate in typically studied populations were highly processed high glycemic load foods … likely attenuating the observed associations between these fats and outcomes.”
In other words, this study found that there was no clear relationship between saturated fat intake and health outcomes when saturated fats replaced refined carbohydrates (sugars, processed carbohydrate sources, etc.). We already know from nutrient substitution studies that replacing saturated fats for refined carbohydrates (and vice versa) has essentially no effect on risk; that is, these are equally harmful. This is uncontroversial, is completely in line with the remaining body of evidence on the topic, and does nothing to refute the original hypothesis about high vs. low intakes. Yet, the study authors still wrote in the Conclusions section that “saturated fats are not associated with all-cause mortality…” To quote the esteemed Florinese cardiovascular researcher and swordsman, Inigo Montoya, “You keep using that word. I do not think it means what you think it means.” This is, under the most charitable light, poor writing. At worst, it is misleading and is not a defensible statement.
9.3 Adjustment
The final issue we will address involves the indirect effect of dietary saturated fats on heart disease risk, by way of influencing blood LDL-cholesterol as the intermediate step. Some studies have instead set out to test the hypothesis that saturated fat has a direct effect on the risk of heart disease, which fails to recognize how plaque development occurs. The way to test such a direct hypothesis involves controlling for all variables aside from saturated fat intake — including blood cholesterol levels. When these studies compare high versus low saturated fat intake, but then adjust for blood LDL-cholesterol, they effectively erase the very mechanism by which dietary saturated fats increase risk. Scarborough 2010 This results in erroneous findings that there is no relationship between dietary saturated fat and heart disease risk. Several commonly cited meta-analyses we have discussed so far suffer from this problem. Siri-Tarino 2010 de Souza 2015
Bottom Line: Animal-derived saturated fats relate to plaque development and heart disease risk indirectly, by altering blood lipoprotein and cholesterol levels as the intermediate step. There is a threshold of effect for risk increases from dietary saturated fats; in particular, when these saturated fats account for greater than 18–20% of total calories. This risk is most significantly impacted when intake is reduced to 10% of calories or less. The foods that replace this saturated fat also have variable effects — whether polyunsaturated fats, plant versus animal-derived monounsaturated fats, complex versus refined carbohydrates, different types of dairy foods, or trans-fats. While several meta-analyses have reported no relationship between saturated fat intake and heart disease risk, these studies commonly suffer from flaws related to 1) inadequate contrast of intake across the “high” vs. “low” threshold to detect a difference, 2) failure to account for the different effects of replacement nutrients, and 3) over-adjustment for blood cholesterol levels, which erases the mediator of the indirect relationship between high saturated fat intake and heart disease, leading to erroneous conclusions.
10 Omega-6 polyunsaturated fats like linoleic acid are unstable and cause inflammation and heart disease
One of the most prevalent beliefs among people on the Internet who debate the significance of blood cholesterol, dietary saturated fats, and other aspects of the material we have presented pertains to polyunsaturated fats. Polyunsaturated fats are a large category of molecules containing multiple double bonds in their chemical structure. These include the well-known “omega 3” (e.g., eicosapentaenoic acid, docosahexaenoic acid, alpha-linolenic acid) and “omega 6” (e.g., linoleic acid) fats, among others.
There are numerous objections to the dietary polyunsaturated fat intake, particularly of the omega-6 linoleic acid. Many of these objections share common threads of reasoning related to things like “inflammation”, oxidation, and chemical “instability.” Concerns about these fats increasing risks of obesity, heart disease, cancer, and other negative health outcomes are frequently expressed as well.
These ideas are typically based on theoretical biochemical mechanisms and pathways in the body or data from studies in rats. When human studies are cited, studies often look at surrogate outcomes, other clinically irrelevant outcomes that are then tied back to a theoretical mechanism, or are drawn from unadjusted ecological data. These are great places to generate hypotheses for further testing in humans. Problems arise when these subsequent studies test those very hypotheses in humans and refute the supposed harms that were theorized mechanisms or rat studies, and people still do not update their beliefs. In fact, they often cling even harder to them.
An example might be a study in humans that shows a change in some blood marker that could then theoretically contribute to disease risk, but when that disease is studied as the main outcome, it is not affected by polyunsaturated fat intake (or may even be improved by it). This is observed repeatedly in this field of research and many others. It is not appropriate to base recommendations to avoid omega 6 polyunsaturated fats on theoretical mechanisms of harm in a petri dish or in rats once we have direct human evidence that does not find the harm to manifest in the real world.
Given the breadth of this topic it is impractical to address every objection here, but we will address a select few.
10.1 Inflammation
This hypothesis invokes the basic biochemical mechanism of omega-6 polyunsaturated fatty acids being metabolized into arachidonic acid, which is then used to create inflammatory molecules known as eicosanoids. Since inflammation can contribute to numerous diseases (including heart disease), this is potentially concerning and is worthy of human study. Fortunately, a systematic review of human studies that have examined the effects of increasing dietary omega-6 linoleic acid intake by 551%, or decreasing it by 90%, found no difference in tissue levels of arachidonic acid. Rett 2011 This is because the biochemical pathways involved are more complex and more highly regulated than the simplistic theory of linoleic acid à arachidonic acid à inflammation would suggest. In fact, radiotracer studies have estimated that humans only convert about 0.2–0.3% of dietary linoleic acid to arachidonic acid, which is a completely insignificant amount. Demmelmair 1999 Hussein 2005 In addition, most dietary sources of polyunsaturated fats (including omega-6 linoleic acid) contain high amounts of antioxidants (such as vitamin E and other polyphenols) that reduce susceptibility to oxidation compared with the antioxidant and polyphenol content of typical saturated fat sources. This is yet another reason why it is important to focus on overall dietary patterns and real-world outcomes, rather than petri dish studies or human studies of unrealistic isolated nutrient supplementation.
To support this, we can look directly at biomarkers of inflammation (like C-reactive protein, mentioned in section 2 above). A systematic review of 15 randomized, controlled trials of linoleic acid feeding concluded that “virtually no evidence is available from randomized, controlled intervention studies among healthy, noninfant human beings to show that addition of linoleic acid to the diet increases the concentration of inflammatory markers.” Johnson 2012 A second meta-analysis of 83 randomized trials including over 41,000 patients examined the long-term effects of higher vs. lower omega-3, omega-6, and total polyunsaturated fat intake on inflammatory bowel disease and biomarkers of inflammation. Ultimately, the researchers found “little or no effect on prevention or treatment of [inflammatory bowel disease] and provides little support for modification of long-term inflammatory status.” In other words, increasing or decreasing these fats had no clear effect on inflammation in human subjects. Ajabnoor 2020 The same results have been found in the context of obesity and fatty liver disease, where a high intake of omega-6 polyunsaturated fats did not cause any signs of inflammation or oxidative stress, but instead reduced liver fat (a good thing) compared with a high saturated fat diet. Bjermo 2012
At this point it no longer matters how many petri dish or rat studies suggest that linoleic acid can increase some inflammatory molecule or cause some other bad outcome. They are all superseded by studies that feed linoleic acid to real humans in varying amounts, which overall find no consistent evidence of increases or decreases in tissue arachidonic acid levels (the supposed mechanism of inflammation), or effects on biomarkers of inflammation. Linoleic acid does not appear to cause inflammation.
10.2 Heart Disease
Many claims around the effects of omega-6 polyunsaturated fats like linoleic acid on heart disease relate to inflammation (addressed above) and risk for oxidation when incorporated into lipoproteins (addressed in section 2 above). At this point we have established that these fats do not cause inflammation in humans, regardless of what theoretical mechanisms or rodent studies are cited. Instead of those types of studies we would prefer to have direct evidence on heart disease outcomes in humans across a wide range of linoleic acid intake. Since humans do not synthesize their own linoleic acid, it must be consumed in the diet — and this means that biomarkers of linoleic acid in the body provide a good reflection of dietary intake.
An analysis of 30 cohort studies including 68,659 people looked at biomarkers of dietary linoleic acid intake and compared these levels with the combined risk of heart attack, stroke, or cardiovascular death. Marklund 2020 The researchers found that higher levels of linoleic acid were associated with lower risk of total cardiovascular disease, and cardiovascular death. Recall that we cannot synthesize linoleic acid, so tissue biomarkers provide a good reflection of dietary intake. Notably, when comparing the lowest tissue linoleic acid levels to the highest, there was not a single cohort that showed a statistically significant increase in heart disease risk. Additionally, higher levels of arachidonic acid were not associated with any increase in heart disease risk – further refuting the original hypothesis.
These findings are consistent with multiple other lines of evidence, such as controlled feeding studies (cited earlier in the article series and discussed in section 9 above) that show substituting polyunsaturated fats with saturated fats results in a decrease in blood cholesterol, apolipoprotein B levels, heart disease risk, and mortality. We have controlled feeding studies, ecological studies, prospective cohort studies, and intervention trials all pointing in the same direction. Clarke 1997 Vartiainen 2010 Jakobsen 2009 Li 2020 Hooper 2020
One often cited exception is the Sydney Diet-Heart Study in the 1960s, which substituted saturated fat for polyunsaturated fat and found higher risk of heart disease. When examining the study more closely, the substituted polyunsaturated fats were from partially hydrogenated margarine products, which at the time contained significant amounts of trans-fats – the type that unequivocally increase heart disease risk and have since been banned from the food supply. This, combined with several other methodological issues, mean the study does not support higher saturated fat intake, nor does it refute the other evidence of non-trans polyunsaturated fats (including linoleic acid) improving risk.
If the theories about omega-6 fats creating arachidonic acid, leading to inflammation and disease were correct, the available human evidence would be pointing in a different direction. Since these collective findings not consistent with that theory, it must be rejected. Although many individuals may report improvements in their health status when eliminating these types of fats from their diet, this is often due to the elimination of ultra-processed junk foods (which often contain vegetable oils), not due to unique harms from the oils themselves. In fact, one meta-analysis of 23 randomized, controlled trials of canola oil intake showed a small decrease in bodyweight when compared with saturated fats. Dehkordi 2019 While the effect was not large, it suggests strongly against the claim that this particular oil is uniquely fattening on its own.
Bottom Line: The consumption of polyunsaturated fats, particularly of omega-6 linoleic acid, is often purported to increase inflammation and increase the risk of cardiovascular disease. Regardless of findings from in vitro or rat studies, when humans are fed polyunsaturated fats (including linoleic acid) in varying quantities, we do not find evidence of increases in inflammatory biomarkers across numerous randomized, controlled trials. Similarly, when observing human outcome trials that substitute polyunsaturated fats (excluding trans-fats) for saturated fats, we observe reliable decreases in blood cholesterol and cardiovascular risk. These findings are supported across multiple lines of evidence, including mechanistic studies, ecological studies, prospective cohort studies, and randomized, controlled intervention trials. Many potentially promising ideas and causal mechanisms around disease are demonstrated in the laboratory or in animal models but fail to produce the same results in humans. This is common and a frequent source of frustration. Human physiology is complex and subject to innumerable variables. What we care about are demonstrable results in real-world outcomes in humans. When those do not materialize, even the tidiest of hypotheses must be let go.
11 How do you explain the increase in rates of heart disease deaths?
When heart disease deaths are viewed for the total population, they do indeed appear to increase. Some people like to take graphs showing increases in overall heart disease deaths, and overlay them with any other variable of choice, such as vegetable oil consumption, to suggest a causal relationship between the two. This is incorrect at best, and deliberately misleading at worst.
When heart disease deaths are calculated as a rate, for example per 100,000 people, rates of heart disease deaths have been dropping for decades. Heart disease deaths have decreased by approximately 72% between 1950 and 2018, and rates of premature death among younger individuals aged 25-64 years have similarly declined by 70% since 1968. CDC 2018 Ritchey 2020 In other words, the observed “increase” is simply due to overall population growth, whereas rates of heart disease deaths are not actually increasing. They are going down.
In addition, the fewer people die from heart disease complications, the more people will remain alive with heart disease present. This may result in an increase in the overall prevalence of heart disease in the living population, while rates of heart disease deaths decline.
We are having successes treating heart disease using the methods detailed in this series of articles. Lowering blood lipids, among other interventions, lowers the risk of cardiovascular disease. This can be accomplished by lifestyle adjustments or medications and both can be very helpful in this regard.
Bottom Line: When heart disease deaths are viewed for the total population, they do indeed appear to increase. This fails to account for the effects of a growing population. When heart disease deaths are calculated as a rate, for example per 100,000 people, rates of heart disease deaths have been dropping steadily for decades.